[This is a slightly revised version of the original blog post, particularly concerning the hP47-α structure; it turned out that there was a small error in the SI.]
Last year I attended for the first time the annual meeting of the German Society for Crystallography (DGK). It took place in Frankfurt/Main from 27-30 March 2023.
It was an interesting event with a broad spectrum of topics. One lecture in particular caught my interest. It was given by by Julia Dshemuchadse (from the Cornell University, Ithaca/NY) with the title:
Simulating complex crystal structures and their assembly in hard and soft condensed materials
Part of the content of the talk has already been published in a very nice paper:
H. Pan & J. Dshemuchadse
Targeted Discovery of Low-Coordinated Crystal Structures via Tunable Particle Interactions
ACS Nano 2023, 17, 7157–7169
https://doi.org/10.1021/acsnano.2c09131
Here, they report on the self-assembly of 20 previously unknown crystal structure types, 14 of which have low coordination numbers (CN), i.e., those between 2 and 7. The coordination numbers were determined with a radial-distrubution function (RDF) approach.
For the 14 new crystal structure types with <CN> = 2 – 7, I recreated the CIFs from the information given in the Supporting Information of the paper (with one exception, see below) and wanted to know: If the crystal structures are hitherto unknown, do some of them build also a hitherto unrecognized net? This seems to be the case. My outcome is as follows:
1. oI4-α
Space group: Imma (No. 74), a = 5.000, b = 5.500, c = 8.750 Å
Atom A: 4e 0.000, 0.250, 0.5329

This crystal structure does not form a net, there are only 2-c vertices.
2. hR3-α
Space group: R-3 (No. 148), a = 5.000, b = 5.000, c = 1.550 Å
Atom A: 3a 0.000, 0.000, 0.000

This structure also does not form a net, there are again only 2-c vertices present.
3. mC32-α
Space group: C2/c (No. 15), a = 5.000, b = 5.000, c = 1.550 Å
Atom A: 8f 0.9364, 0.0010, 0.5622 CN = 2 (light-blue)
Atom B: 8f 0.6882, 0.7457, 0.0580 CN = 2 (light-blue)
Atom C: 8f 0.9398, 0.4528, 0.3197 CN = 3 (dark-blue)
Atom D: 8f 0.8072, 0.2995, 0.1605 CN = 3 (dark-blue)

The light-blue atoms are only 2-coordinated, the dark-blue ones 3-coordinated. After removing the 2-c vertices, which means that the dark-blue ones form a continuous 3-c net, the outcome of the classification with ToposPro is a binodal (3,3)-c net with stoichiometry (3-c)(3-c). The topological type is:
fsg/P m m m –> P b c n (2c,2a,2b; 1/2,0,0); Bond sets: 2,5,6,7:fsg (binodal.ttd) {8^3}
VS [8.8.8(3)] [8.8.8]
This means that it is a subnet of the known fsg net (a binodal (4,6)-c net) and that it can be obtained by transforming the original structure/net in a group-supergroup relationship to the space group Pbcn accompanied with an origin shilft of 0.5 along a and by removing all bonds except the set 2,5,6, and 7. The most important message of this output is, however, that the topology has not yet been found in crystal structures.
4. cI52-α
Space group: Im-3 (No. 204), a = b = c = 8.000 Å
Atom A: 12e 0.1985, 0.0000, 0.5000 CN = 2 (green)
Atom B: 16f 0.6864, 0.6864, 0.6864 CN = 4 (orange)
Atom C: 24g 0.0000, 0.7017, 0.8828 CN = 4 (orange)

The structure is characterized by pentagonal-dodecahedral cages (here shown in orange) located at the origin (0,0,0) and in the center of the unit cell (0.5, 0.5, 0.5) and therefore is related to the structure of chlathrate type-I that is realized, for instance, in cP54-K4Si23 (space group Pm-3n, No. 223). While these cages are occupied with K in cP54-K4Si23, they are empty for cI52-α. Additionally, cP54-K4Si23 contains another six tetradecahedron-shaped cages (24 vertices, 14 faces) with occupied cage centers, while cI52-α contains no additional cages. Instead, the dodecahedron-shaped cages are linked through 2-coordinated species (shown here in green).
As a 2-c vertex is only an edge, they are removed during the simplification procedure of the topological analysis. The result of the classification with ToposPro is:
It is a hitherto unknown binodal (4,4)-c net.
5. tI32-α
Space group: I41/acd (No. 142), a = b = 8.000, c = 6.400 Å
Atom A: 16e 0.2500, 0.1747, 0.1250 CN = 4 (orange)
Atom B: 16f 0.9217, 0.9217, 0.2500 CN = 3 (blue)

The topological analysis shows that this is an already known 3-periodic binodal (3,4)-c net, which is stored in the ToposPro database under the entry 3,4T261. With the help of the online tool topcryst (a kind of front end for ToposPro, accessible via the browser), you can not only carry out the topological analysis, but also look up occurences of strctures that have this underlying net, but the number of occurences here is indeed zero.
6. hP44-α
Space group: P6/mmm (No. 191), a = b = 8.000, c = 8.080 Å
Atom A: 2d 0.33333, 0.66667, 0.50000 CN = 0 (pink)
Atom B: 2e 0.00000, 0.00000, 0.28520 CN = 0 (pink)
Atom C: 4h 0.33333, 0.66667, 0.11550 CN = 4 (orange)
Atom D: 6k 0.26650, 0.00000, 0.50000 CN = 4 (orange)
Atom E: 6l 0.13470, 0.26940, 0.00000 CN = 4 (orange)
Atom F: 12n 0.38270, 0.00000, 0.30970 CN = 4 (orange)
Atom G: 12o 0.21020, 0.42040, 0.81160 CN = 4 (orange)

The framework of hP44-α is identical with that of the clathrate type IV with a unit cell containing three 20-vertex pentagonal dodecahedron-, two 24-vertex tetradecahedron-, and two 26-vertex pentadecahedron-shaped cages. However, the hP44-α structure is missing one set of cage centers at Wyckoff position 3f, corresponding to the (smallest) cage centers of the pentagonal-dodecahedron cages.
Not unexpectedly, the underlying net is already known: It is the 5-nodal (4,4,4,4,4)-c net know as zra-d.
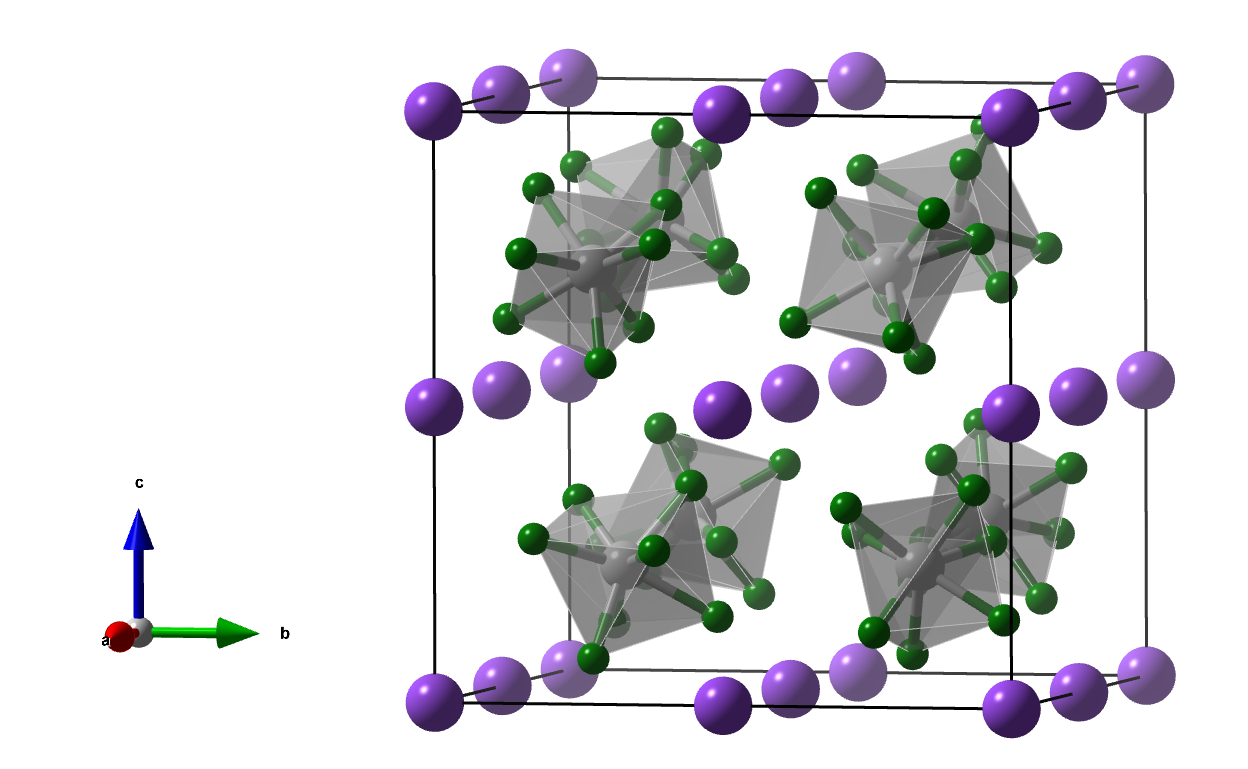
7. hP47-α
Initially, I was not able to infer the given coordination numbers on the basis of the given specificaton of the crystal structure in the SI. After contacting Julia, she was so kind to send me the CIF file and it turned out that there was a small mistake in the SI. The correct specification is as follows:
Space group: P6/mmm (No. 191), a = b = 5.15931, c = 5.23651 Å
Atom A: 2d 0.33333, 0.66667, 0.50000 CN = 0 (pink)
Atom B: 2e 0.00000, 0.00000, 0.70510 CN = 0 (pink)
Atom C: 4h 0.66667, 0.33333, 0.88480 CN = 5.5 (black)
Atom D: 6k 0.69810, 0.00000, 0.50000 CN = 2 (light-blue)
Atom E: 6l 0.87383, 0.74766, 0.00000 CN = 4 (orange)
Atom F: 6l 0.45933, 0.91865, 0.00000 CN = 2, Occ = 0.5 (light-blue)
Atom G: 12n 0.62722, 0.62722, 0.68902 CN = 4 (orange)
Atom H: 12o 0.20991, 0.41982, 0.18906 CN = 4 (orange)


Removing the 0- and 2-c vertices (this implies also a reduction of the CN of the 4h site from 5.5 to 4) leads to the already known 4-nodal (4,4,4,4)-c net doh.
8. cF144-Na8Si136
Space group: Fd-3m (No. 227), a = b = c = 11.0000 Å
Atom A: 8a 0.0000, 0.0000, 0.0000 CN = 0
Atom B: 8b 0.5000, 0.5000, 0.5000 CN = 4
Atom C: 32e 0.4074, 0.4074, 0.4074 CN = 4
Atom D: 96g 0.4393, 0.4393, 0.2513 CN = 4

The cF144-Na8Si136 structure is strictly speaking not a new crystal structure type that has been discovered in the work of Pan & Dshemuchadse but has already been observed experimentally [1,2]. It corresponds to the clathrate type II structure of cF160-Na24Si136 with missing cage centers within the pentagonal dodecahedral cages, whereas the centers of the hexadecahedron-shaped cages are occupied in both structures.
The underlying net is also already known. It is the trinodal (4,4,4)-c net mtn, very well known in the zeolite community; it is the net of the zeolite ZSM-39.
9. mC32-β
Space group: C2/c (No. 15), a = 7.0000 b = 6.3700, c = 4.6900 Å, β = 132°
Atom A: 4e 0.0000, 0.7746, 0.2500 CN = 4 (orange)
Atom B: 4e 0.0000, 0.3679, 0.2500 CN = 4 (orange)
Atom C: 8f 0.2901, 0.0670, 0.4208 CN = 3 (green)
Atom D: 8f 0.0843, 0.2798, 0.5821 CN = 4 (orange)
Atom E: 8f 0.1184, 0.0797, 0.7191 CN = 5 (purple)

Here, we have again a hitherto unknown net! It is a 5-nodal (3,4,4,4,5)-net.
10. cP8-α
Space group: Pn-3m (No. 224), a = b = c = 5.000 Å
Atom A: 8e 0.1375, 0.1375, 0.1375 CN = 4

Nothing special here: The underlying net of this structure is very well known, it is the augmented version of diamond net, the uninodal 4-c net dia-d.
11. mC12-α
Space group: C2/m (No. 12), a = 8.000, b = 4.000, c = 5.360 Å, β = 107.4°
Atom A: 4i 0.0761, 0.0000, 0.1724 CN = 4 (orange)
Atom B: 4i 0.3495, 0.0000, 0.4541 CN = 4 (orange)
Atom C: 4i 0.5018, 0.0000, 0.1967 CN = 5 (blue)
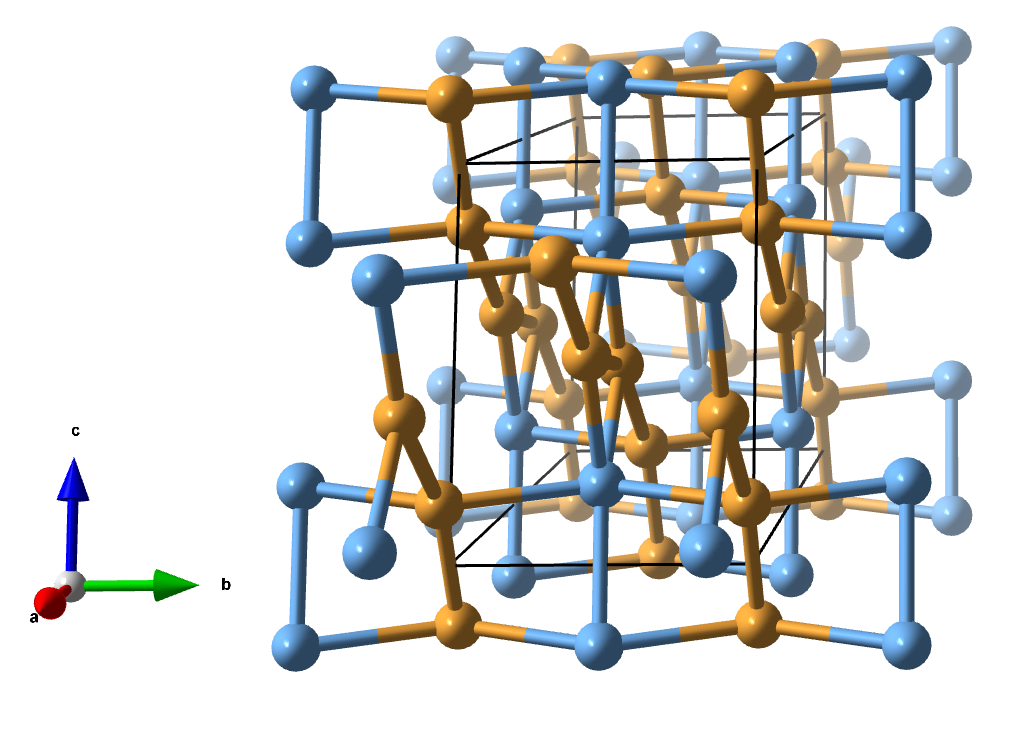
The underlying net of this crystal structure is again new! It is a trinodal (4,4,5)-c net.
12. hP22-α
Space group: P63/mcm (No. 193), a = b = 7.000, c = 7.470 Å
Atom A: 4d 0.3333, 0.6667, 0.0000 CN = 6 (pink)
Atom B: 6g 0.1673, 0.0000, 0.2500 CN = 4 (blue)
Atom C: 12k 0.4258, 0.0000, 0.1149 CN = 5 (green)
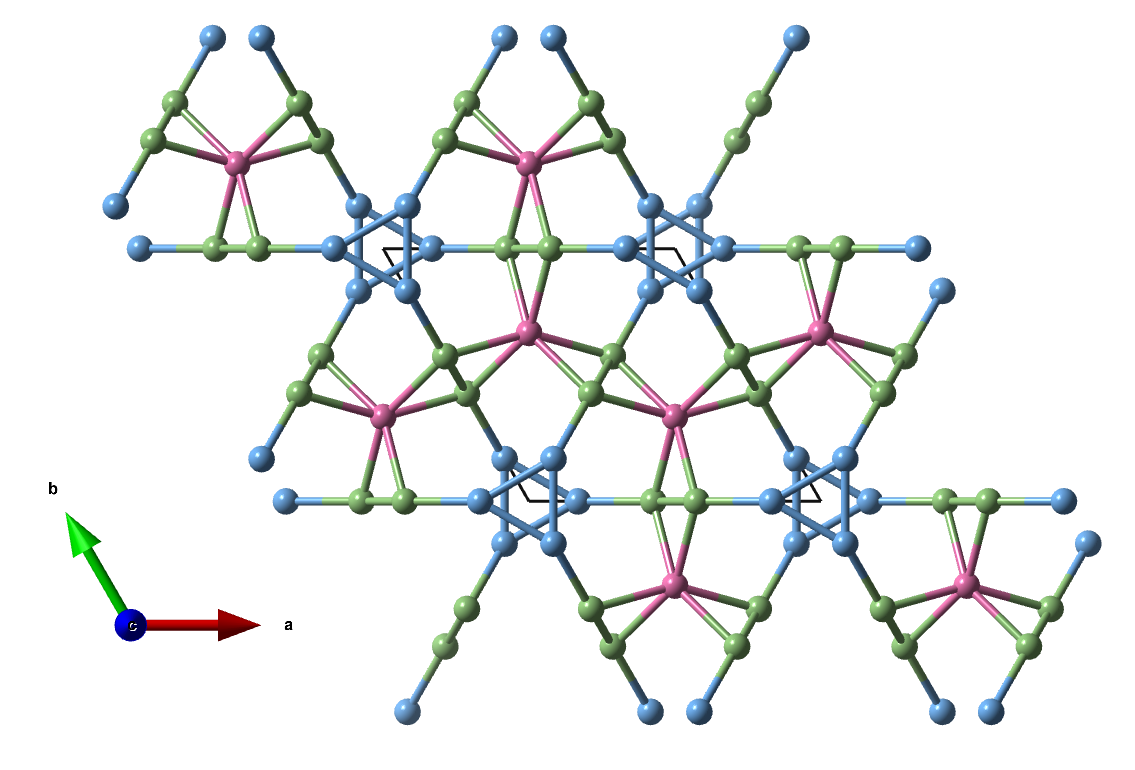
Here we have another hitherto unknown net, a trinodal (4,5,6)-c net.
13. tI16-α
Space group: I41/amd (No. 141), a = b = 4.500, c = 9.000 Å
Atom A: 8d 0.0000, 0.2500, 0.6250 CN = 6 (blue)
Atom B: 8e 0.0000, 0.0000, 0.1032 CN = 7 (golden)

This net is also new, a hitherto unknow binodal (6,7)-c net.
14. cI48-α
Space group: I-43d (No. 220), a = b = c = 10.000 Å
Atom A: 48e 0.0314, 0.8993, 0.0467 CN = 7 (blue)

Finally, we have another structure in which the underlying network is already known, it is the uninodal 7-c net called svx.
Altogether, out of these 14 new crystal structure types, 5 constitute an underlying network that was previously unknown.